Lennard-Jones fluid¶
The very basics of LAMMPS through a simple example
The objective of this tutorial is to perform simple MD simulations using LAMMPS. The system consists of a Lennard-Jones fluid composed of neutral particles with two different effective diameters, contained within a cubic box with periodic boundary conditions. In this tutorial, basic MD simulations in the microcanonical (NVE) and canonical (NVT) ensembles are performed, and basic quantities are calculated, including the potential and kinetic energies.
This tutorial is compatible with the 22Jul2025 LAMMPS version.
Cite
If you find these tutorials useful, you can cite A Set of Tutorials for the LAMMPS Simulation Package [Article v1.0] by Simon Gravelle, Cecilia M. S. Alvares, Jacob R. Gissinger, and Axel Kohlmeyer, published in LiveCoMS, 6(1), 3037 (2025) [14].
My first input¶
To run a simulation using LAMMPS, you need to write an input script
containing a series of commands for LAMMPS to execute, similar to Python
or Bash scripts. For clarity, the input scripts for this tutorial will
be divided into five categories, which will be filled out step by step.
To set up this tutorial using LAMMPS graphical user interface
(LAMMPS–GUI), select Start LAMMPS Tutorial 1
from the Tutorials menu and follow the instructions. This will
select (or create, if needed) a folder, place the initial input
file initial.lmp in it, and open the file in the LAMMPS–GUI Editor window:
# PART A - ENERGY MINIMIZATION
# 1) Initialization
# 2) System definition
# 3) Settings
# 4) Monitoring
# 5) Run
If you are not using LAMMPS-GUI
All tutorials can be followed without using LAMMPS-GUI. To do so, create a new folder and add a file named initial.lmp inside it. Open the file in a text editor of your choice and copy the previous lines into it.
Everything that appears after a hash symbol (#) is a comment
and ignored by LAMMPS. These five categories are not required in every input script an do not
necessarily need to be in that exact order. For instance, the Settings
and the Monitoring categories could be inverted, or
the Monitoring category could be omitted. However, note that
LAMMPS reads input files from top to bottom and processes each command
immediately. Therefore, the Initialization and
System definition categories must appear at the top of the
input, and the Run category must appear at the bottom. Also, the
specifics of some commands can change after global settings are modified, so the
order of commands in the input script is important.
Initialization¶
In the first section of the script, called Initialization,
global parameters for the simulation are defined, such as units, boundary conditions
(e.g., periodic or non-periodic), and atom types (e.g., uncharged point particles
or extended spheres with a radius and angular velocities). These commands must be
executed before creating the simulation box or they will cause
an error. Similarly, many LAMMPS commands may only be
entered after the simulation box is defined. Only a limited
number of commands may be used in both cases. Update the initial.lmp file
so that the Initialization section appears as follows:
# 1) Initialization
units lj
dimension 3
atom_style atomic
boundary p p p
Note
Strictly speaking, none of the four commands specified in the
Initialization section are mandatory, as they correspond to the
default settings for their respective global properties. However,
explicitly specifying these defaults is considered good practice to
avoid confusion when sharing input files with other LAMMPS users.
The first line, units lj, specifies the use of reduced
units, where all quantities are dimensionless. This unit system is a
popular choice for simulations that explore general statistical
mechanical principles, as it emphasizes relative differences between
parameters rather than representing any specific material. The second
line, dimension 3, specifies that the simulation is conducted
in 3D space, as opposed to 2D, where atoms are confined to move only in
the xy-plane. The third line, atom_style atomic, designates
the atomic style for representing simple, individual point particles.
In this style, each particle is treated as a point with a mass, making
it the most basic atom style. Other atom styles can incorporate
additional attributes for atoms, such as charges, bonds, or molecule
IDs, depending on the requirements of the simulated model. The last
line, boundary p p p, indicates that periodic boundary
conditions are applied along all three directions of space, where the
three p stand for \(x\), \(y\), and \(z\), respectively.
Alternatives are fixed non-periodic (f), shrink-wrapped non-periodic (s), and
shrink-wrapped non-periodic with minimum (m). For non-periodic
boundaries, different options can be assigned to each dimension, making
configurations like boundary p p fm valid for systems such as
slab geometries.
Note
Each LAMMPS command is accompanied by extensive online documentation
that lists and discusses the different options for that command.
Most LAMMPS commands also have default settings that are applied if no
value is explicitly specified. The defaults for each com-
mand are listed at the bottom of its documentation page.
From the LAMMPS–GUI editor buffer, you can access the documentation by
right-clicking on a line containing a command (e.g., units lj)
and selecting View Documentation for `units'. This action
should prompt your web browser to open the corresponding URL for the
online manual.
Note
Most LAMMPS commands have default settings that are applied if no value is explicitly specified. The defaults for each command are listed at the bottom of its documentation page.
System definition¶
The next step is to create the simulation box and populate it with
atoms. Modify the System definition category of
initial.lmp as shown below:
# 2) System definition
region simbox block -20 20 -20 20 -20 20
create_box 2 simbox
create_atoms 1 random 1500 34134 simbox overlap 0.3
create_atoms 2 random 100 12756 simbox overlap 0.3
The first line, region simbox (...), defines a region named
simbox that is a block (i.e., a rectangular cuboid) extending
from -20 to 20 units along all three spatial dimensions. The second
line, create_box 2 simbox, initializes a simulation box based
on the region simbox and reserves space for two types of atoms.
In LAMMPS, every atom is assigned an atom type property. This property selects which force
field parameters (here, the Lennard-Jones parameters, \(\epsilon_{ij}\)
and \(\sigma_{ij}\)) are applied to each pair of atoms via the pair_coeff
command (see below). We discuss in Pulling on a carbon nanotube how this applies to
many-body pair styles, and in Polymer in water how this applies to
Coulomb interactions.
Note
From this point on, the number of atom types is locked in, and any command referencing an atom type larger than 2 will trigger an error. While it is possible to allocate more atom types than needed, you must assign a mass and provide force field parameters for each atom type. Failing to do so will cause LAMMPS to terminate with an error.
The third line, create_atoms (...), generates 1500 atoms of
type 1 at random positions within the simbox region.
The integer 34134 is a seed for the internal random number generator, which
can be changed to produce different sequences of random numbers and,
consequently, different initial atom positions. The fourth line adds
100 atoms of type 2. Both create_atoms commands use the
optional argument overlap 0.3, which enforces a minimum
distance of 0.3 length units between the randomly placed atoms. This
constraint helps avoid close contacts between atoms, which can lead
to excessively large forces and simulation instability.
Each created atom in LAMMPS is automatically
assigned a unique atom ID, which serves as a numerical
identifier to distinguish individual atoms throughout the
simulation. Atom IDs by default have the range from 1 to
the total number of atoms, but this is not enforced.Deleting atoms, for example,
causes holes in the list of atom IDs.
Note
Another way to define a system in LAMMPS, besides the
create_atoms commands, is to import an existing topology
file containing atomic coordinates as well as, optionally, other
attributes such as atomic velocities and the force field parameters
using the read_data command, as shown in Pulling on a carbon nanotube.
Looking for help with your project ?
Get guidance for your LAMMPS simulations and receive personalized advice for your project.
Settings¶
Next, we specify the settings for the two atom types. Modify the
Settings category of initial.lmp as follows:
# 3) Settings
mass 1 1.0
mass 2 5.0
pair_style lj/cut 4.0
pair_coeff 1 1 1.0 1.0
pair_coeff 2 2 0.5 3.0
The two mass commands assign a mass of 1.0 and 5.0 units to the
atoms of type 1 and 2, respectively. The third line,
pair_style lj/cut 4.0, specifies that the atoms will be
interacting through a Lennard-Jones (LJ) potential with a cut-off equal
to \(r_c = 4.0\) length units [15, 16]:
where \(r\) is the inter-particle distance, \(\epsilon_{ij}\) is
the depth of the potential well that determines the interaction strength, and
\(\sigma_{ij}\) is the distance at which the potential energy equals zero.
The indices \(i\) and \(j\) refer to pairs atoms with the corresponding atom types.
The fourth line, pair_coeff 1 1 1.0 1.0, specifies the
Lennard-Jones coefficients for interactions between pairs of atoms
that both have atom type 1: the energy parameter \(\epsilon_{11} = 1.0\) and
the distance parameter \(\sigma_{11} = 1.0\). Similarly, the last line
sets the Lennard-Jones coefficients for interactions between atoms
of type 2, \(\epsilon_{22} = 0.5\), and \(\sigma_{22} = 3.0\).
Note
By default, LAMMPS calculates the mixed force field coefficients for different
atom types using geometric averages: \(\epsilon_{ij} = \sqrt{\epsilon_{ii} \epsilon_{jj}}\),
\(\sigma_{ij} = \sqrt{\sigma_{ii} \sigma_{jj}}\). In the present case,
\(\epsilon_{12} = \sqrt{1.0 \times 0.5} = 0.707\), and
\(\sigma_{12} = \sqrt{1.0 \times 3.0} = 1.732\). Other rules
can be selected using the pair_modify command.
Single-point energy¶
The system is now fully parameterized. Let us complete the two remaining categories,
Monitoring and Run, by adding the following lines
to initial.lmp:
# 4) Monitoring
thermo 10
thermo_style custom step etotal press
# 5) Run
run 0 post no
The thermo 10 command instructs LAMMPS to print thermodynamic
information to the console every specified number of steps, in this case,
every 10 simulation steps. The thermo_style custom command
defines the specific outputs, which in this case are the step number
(step), total energy \(E\) (etotal), and pressure \(p\) (press).
The run 0 post no command instructs LAMMPS to initialize forces and energy
without actually running the simulation. The post no option disables
the post-run summary and statistics output.
Note
The thermodynamic information printed by LAMMPS
using thermo_style custom keywords refers to instanta-
neous values of the specified thermodynamic properties
at each output step, not cumulative averages. However,
LAMMPS also allows to reference a wide variety of custom data from
compute styles, fix styles, and variables.
These can be used for on-the-fly analysis, including cumulative and sliding-window averages.
You can now run LAMMPS (basic commands for running LAMMPS are provided in Running LAMMPS). The simulation should finish quickly.
With the default settings, LAMMPS–GUI will open two windows: one
displaying the console output and another with a chart. The Output window
will display information from the executed commands, including the
total energy and pressure at step 0,
as specified by the thermodynamic data request. Since no actual simulation
steps were performed, the Charts window will be empty.
Snapshot image – At this point, you can create a snapshot image of the current system
using the Image Viewer window, which can be accessed by
clicking the Create Image button in the Run menu. The
image viewer works by instructing LAMMPS to render an image of the
current system using its internal rendering library via the dump image
command. The resulting image is then displayed, with various
buttons available to adjust the view and rendering style. This will always
capture the current state of the system.
Energy minimization¶
Now, replace the run 0 post no command line with the
following minimize command:
# 5) Run
minimize 1.0e-6 1.0e-6 1000 10000
This tells LAMMPS to perform an iterative energy minimization of the system. Specifically, LAMMPS will compute the forces on all atoms and then update their positions according to a selected algorithm, aiming to reduce the potential energy. By default, LAMMPS uses the conjugate gradient (CG) algorithm [17]. The simulation will stop as soon as one of the four minimizer criteria is met. LAMMPS will then report which stopping criterion was satisfied, along with selected system properties at both the initial and final steps. Note that, except for trivial systems, minimization algorithms will find a local minimum rather than the global minimum.
Run the minimization and observe that LAMMPS-GUI captures the output
and updates the chart in real time. This run executes quickly (depending
on your computer’s capabilities), and thus LAMMPS-GUI may fail to capture some
of the thermodynamic data. In that
case, use the Preferences dialog to reduce the data update
interval and switch to single-threaded, unaccelerated execution in the
Accelerators tab. You can repeat the run; each new attempt will start
fresh, resetting the system and re-executing the script from the beginning.
Run the minimization. The potential energy, \(U\), decreases from a positive value to a negative value (as can also be seen in the figure below). Note that during energy minimization, the potential energy equals the total energy of the system, \(E = U\), since the kinetic energy, \(K\), is zero. The initially positive potential energy is expected, as the atoms are created at random positions within the simulation box, with some in very close proximity to each other. This proximity results in a large initial potential energy due to the repulsive branch of the Lennard-Jones potential [i.e., the term \(1/r^{12}\) in Eq. (1)]. As the energy minimization progresses, the energy decreases - first rapidly - then more gradually, before plateauing at a negative value. This indicates that the atoms have moved to reasonable distances from one another.
Create and save a snapshot image of the simulation state after the minimization, and compare it to the initial image. You should observe that the atoms are clumping together as they move toward positions of lower potential energy.
Molecular dynamics¶
After energy minimization, any overlapping atoms are displaced, and
the system is ready for a molecular dynamics simulation. To continue
from the result of the minimization step, append the MD simulation
commands to the same input script, initial.lmp. Add the
following lines immediately after the minimize command:
# PART B - MOLECULAR DYNAMICS
# 4) Monitoring
thermo 50
thermo_style custom step temp etotal pe ke press
Since LAMMPS reads inputs from top to bottom, these lines will
be executed after the energy minimization. Therefore,
there is no need to re-initialize or re-define the
system. The thermo command is called a second time to
update the output frequency from 10 to 50 as soon as PART B of
the simulation starts. In addition, a new thermo_style
command is introduced to specify the thermodynamic information LAMMPS should
print during PART B. This adjustment is made because, during
molecular dynamics, the system exhibits a non-zero temperature \(T\) (and
consequently a non-zero kinetic energy \(K\), keyword ke), which are useful to monitor.
The pe keyword represents the potential energy of the system, \(U\), such that
\(U + K = E\).
Then, add a second Run category by including the following
lines in PART B of initial.lmp:
# 5) Run
fix mynve all nve
timestep 0.005
run 50000
The fix nve command updates the positions and velocities of the
atoms in the group all at every step. More specifically, this command integrates
Newton’s equations of motion using the velocity-Verlet algorithm. The group all
is a default group that contains all atoms. The last two lines specify
the value of the timestep and the number of steps for the
run, respectively, for a total duration of 250 time units.
Note
Since the only command affecting forces and velocities in the
present script is fix nve, and periodic boundary conditions are applied
in all directions, the MD simulation will be performed in the microcanonical (NVE) ensemble, which
maintains a constant number of particles and a fixed box volume. In
this ensemble, the system does not exchange energy with anything
outside the simulation box.
Run the simulation using LAMMPS. Initially, the system is not equilibrated, as the potential energy decreases while the kinetic energy increases. After approximately 40000 steps, the values for both kinetic and potential energy plateau, indicating that the system has reached equilibrium, with the total energy fluctuating around a certain constant value.
Now, we change the second Run section to (note the smaller number of
MD steps):
# 5) Run
fix mynve all nve
fix mylgv all langevin 1.0 1.0 0.1 10917
timestep 0.005
run 15000
The new command adds a Langevin thermostat to the atoms in the group
all, with a target temperature of 1.0 temperature units
throughout the run (the two numbers represent the target temperature at
the beginning and at the end of the run, which results in a temperature
ramp if they differ) [18]. A damping
parameter of 0.1 is used. It determines how rapidly the temperature is
relaxed to its desired value. In a Langevin thermostat, the atoms are
subject to friction and random noise (in the form of randomly added
velocities). Since a constant friction term removes more kinetic energy
from fast atoms and less from slow atoms, the system will eventually
reach a dynamic equilibrium where the kinetic energy removed and added
are about the same. The number 10917 is a seed used to initialize the
random number generator used inside of fix langevin; you can
change it to perform statistically independent simulations. In the
presence of a thermostat, the MD simulation will be performed in the
canonical or NVT ensemble.
Run the simulation again using LAMMPS. From the information printed in the log file, one can see that the temperature starts from 0 but rapidly reaches the requested value and stabilizes itself near \(T=1\) temperature units. One can also observe that the potential energy, \(U\), rapidly decreases during energy minimization (see the figure below). After the molecular dynamics simulation starts, \(U\) increases until it reaches a plateau value of about -0.25. The kinetic energy, \(K\), is equal to zero during energy minimization and then increases rapidly during molecular dynamics until it reaches a plateau value of about 1.5.
Note
All simulations presented in these tutorials are deliberately kept short so they can be executed on a personal computer. These runs are not intended to produce statistically meaningful results, and should not be considered suitable for publication (see for instance Ref. [19]).
From the information
printed in the Output window, one can see that the temperature
starts from 0 but rapidly reaches the requested value and
stabilizes itself near \(T=1\) temperature units. One can also observe that
the potential energy, \(U\), rapidly decreases during energy
minimization (see the figure below). After
the molecular dynamics simulation starts, \(U\) increases until
it reaches a plateau value of about -0.25. The kinetic energy,
\(K\), is equal to zero during energy minimization and then
increases rapidly during molecular dynamics until it reaches
a plateau value of about 1.5.
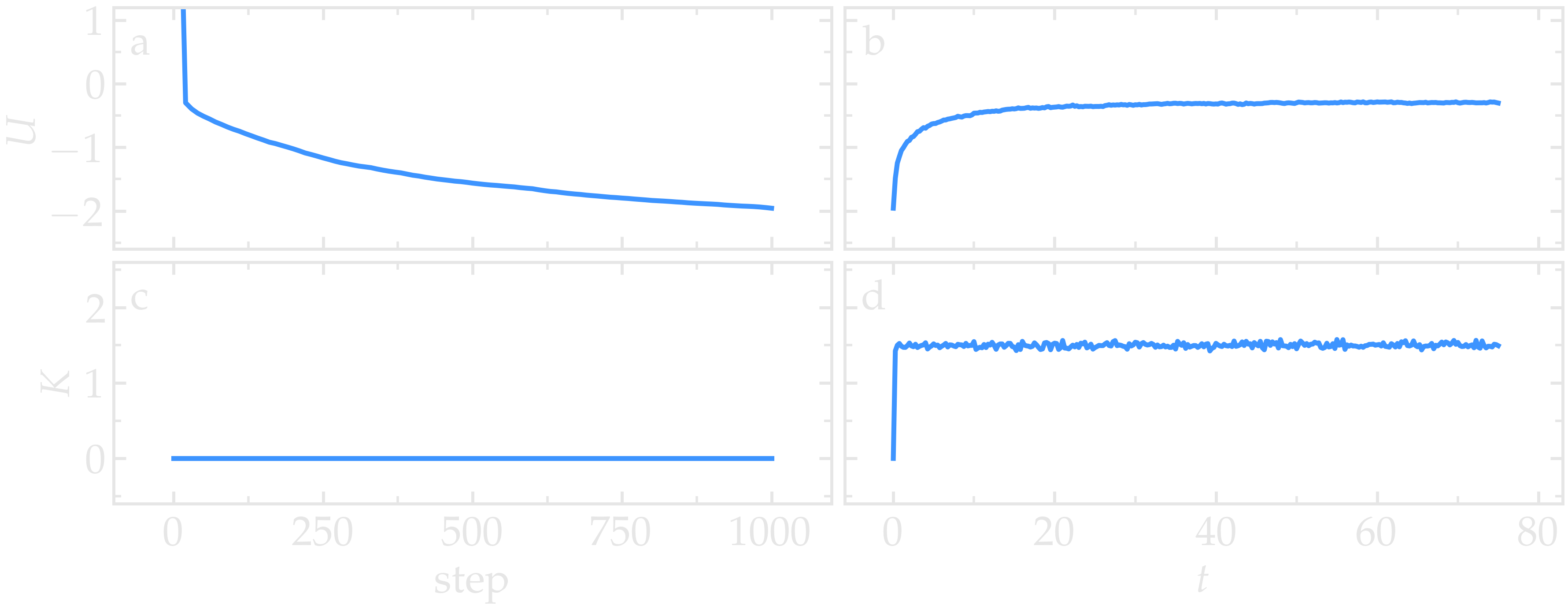
Figure: (a) Potential energy, \(U\), of the binary mixture as a function of the step during energy minimization. (b) Potential energy, \(U\), as a function of time, \(t\), during molecular dynamics in the NVT ensemble. (c) Kinetic energy, \(K\), during energy minimization. (d) Kinetic energy, \(K\), during molecular dynamics.
Trajectory visualization¶
So far, the simulation has been mostly monitored through the analysis of thermodynamic information. To better follow the evolution of the system and visualize the trajectories of the atoms, let us print the positions of the atoms in a file at a regular interval.
Add the following command to the Monitoring section
of PART B of the initial.lmp file:
dump mydmp all atom 100 dump.lammpstrj
Run the initial.lmp file using LAMMPS again. A file named dump.lammpstrj must appear alongside initial.lmp. The .lammpstrj file can be opened using VMD [12] or OVITO [13].
Use the dump image command to create snapshot images during the simulation. We
have already explored the Image Viewer window. Open it again
and adjust the visualization to your liking using the available buttons.
Now you can copy the commands used to create this visualization to the
clipboard by either using the Ctrl-D keyboard shortcut or
selecting Copy dump image command from the File menu.
This text can be pasted into the Monitoring section
of PART B of the initial.lmp file. This may look like
the following:
dump viz all image 100 myimage-*.ppm type type size 800 800 zoom 1.452 shiny 0.7 fsaa yes &
view 80 10 box yes 0.025 axes no 0.0 0.0 center s 0.483725 0.510373 0.510373
dump_modify viz pad 9 boxcolor royalblue backcolor white adiam 1 1.6 adiam 2 4.8
The $&$ is used to continue the command on a new line.
These commands tell LAMMPS to generate NetPBM format images every 100
steps. The two type keywords are for color and
diameter, respectively. Run the initial.lmp using
LAMMPS again, and a new window named Slide Show will pop up.
It will show each image created by the dump image as it is
created. After the simulation is finished (or stopped), the slideshow
viewer allows you to animate the trajectory by cycling through the
images. The window also allows you to export the animation to a movie
(provided the FFMpeg program is installed) and to bulk delete those
image files.
The rendering of the system can be further adjusted using the many
options of the dump image command. For instance, the value for the
shiny keyword is used to adjust the shininess of the atoms, the
box keyword adds or removes a representation of the box, and
the view and zoom keywords adjust the camera (and so on).
Improving the script¶
Let us improve the input script and perform more advanced operations, such as specifying initial positions for the atoms and restarting the simulation from a previously saved configuration.
Control the initial atom positions¶
Open the improved.min.lmp, which was downloaded during the
tutorial setup. This file contains the Part A of the
initial.lmp file, but without any
commands in the System definition section:
# 1) Initialization
units lj
dimension 3
atom_style atomic
boundary p p p
# 2) System definition
# 3) Settings
mass 1 1.0
mass 2 10.0
pair_style lj/cut 4.0
pair_coeff 1 1 1.0 1.0
pair_coeff 2 2 0.5 3.0
# 4) Monitoring
thermo 10
thermo_style custom step etotal press
# 5) Run
minimize 1.0e-6 1.0e-6 1000 10000
We want to create the atoms of types 1 and 2 in two separate
regions. To achieve this, we need to add two region commands and then
reintroduce the create_atoms commands, this time using the new
regions instead of the simulation box region to place the atoms:
# 2) System definition
region simbox block -20 20 -20 20 -20 20
create_box 2 simbox
# for creating atoms
region cyl_in cylinder z 0 0 10 INF INF side in
region cyl_out cylinder z 0 0 10 INF INF side out
create_atoms 1 random 1000 34134 cyl_out
create_atoms 2 random 150 12756 cyl_in
The side in and side out keywords are used to define
regions representing the inside and outside of the cylinder of radius
10 length units. Then, append a sixth section titled Save system at the end
of the file, ensuring that the write_data command is placed after
the minimize command:
# 6) Save system
write_data improved.min.data
Note
A key improvement to the input is the addition of the
write_data command. This command writes the state of the
system to a text file called improved.min.data. This
.data file will be used later to restart the simulation from
the final state of the energy minimization step, eliminating the need
to repeat the system creation and minimization.
Run the improved.min.lmp file using LAMMPS–GUI. At the end of the simulation, a file called improved.min.data is created.
You can view the contents of improved.min.data from LAMMPS–GUI, by
right-clicking on the file name in the editor and selecting the entry
View file improved.min.data.
The created .data file contains all the information necessary
to restart the simulation, such as the number of atoms, the box size,
the masses, and the pair coefficients. This .data file also
contains the final positions of the atoms, along with their IDs and types, within the Atoms
section. The first five columns of the Atoms section
correspond (from left to right) to the atom indexes (from 1 to the total
number of atoms, 1150), the atom types (1 or 2 here), and the positions
of the atoms \(x\), \(y\), \(z\). The last three columns are image flags that
keep track of which atoms crossed the periodic boundary. The exact
format of each line in the Atoms section depends on the choice
of atom_style, which determines which per-atom data is set and
stored internally in LAMMPS.
Note
Instead of the write_data command, you can also use the
write_restart command to save the state
of the simulation to a binary restart file. Binary restart files are
more compact, faster to write, and contain more information, making them often
more convenient to use. For example, the choice of atom_style
or pair_style is recorded, so those commands do not need to be issued
before reading the restart. Note however that restart files are not expected to be
portable across LAMMPS versions or platforms. Therefore, in these tutorials,
and with the exception of Tutorial 3, Polymer in water,
we primarily use write_data to provide you with a reference
copy of the data file that works regardless of your LAMMPS version and platform.
Restarting from a saved configuration¶
To continue a simulation from the saved configuration, open the
improved.md.lmp file, which was downloaded during the tutorial setup.
This file contains the Initialization part from initial.lmp
and improved.min.lmp:
# 1) Initialization
units lj
dimension 3
atom_style atomic
boundary p p p
# 2) System definition
# 3) Settings
# 4) Monitoring
# 5) Run
Since we read most of the information from the data file, we don’t need
to repeat all the commands from the System definition
and Settings categories. The exception is the pair_style
command, which now must come before the simulation box is defined,
meaning before the read_data command. Add the following
lines to improved.md.lmp:
# 2) System definition
pair_style lj/cut 4.0
read_data improved.min.data
By visualizing the system, you may
have noticed that some atoms left their original region during
minimization. To start the simulation from a clean slate, with only
atoms of type 2 inside the cylinder and atoms of type 1 outside the
cylinder, let us delete the misplaced atoms by adding the following
commands to the System definition category to improved.md.lmp:
region cyl_in cylinder z 0 0 10 INF INF side in
region cyl_out cylinder z 0 0 10 INF INF side out
group grp_t1 type 1
group grp_t2 type 2
group grp_in region cyl_in
group grp_out region cyl_out
group grp_t1_in intersect grp_t1 grp_in
group grp_t2_out intersect grp_t2 grp_out
delete_atoms group grp_t1_in
delete_atoms group grp_t2_out
The first two region commands recreate the previously defined
regions, which is necessary since regions are not saved by the
write_data command. The first two group commands
create groups containing all the atoms of type 1 and all the
atoms of type 2, respectively. The next two group commands
create atom groups based on their positions at the beginning of the
simulation, i.e., when the commands are being read by LAMMPS. The last
two group commands create atom groups based on the intersection
between the previously defined groups. Finally, the two
delete_atoms commands delete the atoms of type 1
located inside the cylinder and the atoms of type 2 located
outside the cylinder, respectively.
Since LAMMPS has a limited number of custom groups (30), it is good practice to delete groups that are no longer needed. This can be done by adding the following four commands to improved.md.lmp:
# delete no longer needed groups
group grp_in delete
group grp_out delete
group grp_t1_in delete
group grp_t2_out delete
Let us monitor the number of atoms of each type inside the cylinder as a function of time by creating the following equal-style variables:
variable n1_in equal count(grp_t1,cyl_in)
variable n2_in equal count(grp_t2,cyl_in)
The equal-style variables are expressions evaluated
during the run and return a number. Here, they are defined to count
the number of atoms of a specific group within the cyl_in region.
Note
The n1_in and n2_in defined above are
equal-style variables, which evaluate a numerical expression using the
count() function. Other common LAMMPS variable types include
atom-style, index, and loop.
In addition to counting the atoms in each region, we will also extract
the coordination number of type 2 atoms around type 1 atoms. The
coordination number measures the number of type 2 atoms near
type 1 atoms, defined by a cutoff distance. Taking the average provides
a good indicator of the degree of mixing in a binary mixture. This
is done using two compute commands: the first counts the
coordinated atoms, and the second calculates the average over all type 1
atoms. Add the following lines to improved.md.lmp:
compute coor12 grp_t1 coord/atom cutoff 2 group grp_t2
compute sumcoor12 grp_t1 reduce ave c_coor12
The compute reduce ave command is used to average the per-atom
coordination number calculated by the coord/atom
compute command. Compute commands do not print or output
anything by themselves, nor are they automatically executed; they
require a consumer command that references the compute. In this case, the
first compute is referenced by the second, and we reference the second
in a thermo_style custom command (see below).
Note
LAMMPS compute commands can produce
a wide variety of data and one can identify the category from the
name of the compute style: global data (no suffix), local data
(local suffix), per-atom data (atom suffix), per-chunk data
(chunk suffix), per-gridpoint data (grid suffix). In the example
above, the compute coord/atom produces per-atom data, which
is used as input for compute reduce which returns global
data. For global data three kinds of data exists: scalars (single
values), vectors (one-dimensional arrays), or arrays
(two-dimensional tables). When referencing results of a compute,
you can use indices: for example, c_mycompute refers to
the entire scalar, vector, or array, and c_mycompute[1]
refers to its first element (in case of vector or array). In some
cases also wildcards like “*” can be used to, for instance, refer to all elements
of a vector instead of having specify all elements individually.
In general, consumer commands (fix styles or dump styles,
variables, or other compute styles) can only work with certain data
types or need to have keywords set to select which data to use.
You need to check the documentation of each command to ensure
compatibility.
Note
There is no need for a Settings
section, as the settings are taken from the .data file.
Finally, let us complete the script by adding the following lines to improved.md.lmp:
# 4) Monitoring
thermo 1000
thermo_style custom step temp pe ke etotal press v_n1_in v_n2_in c_sumcoor12
dump viz all image 1000 myimage-*.ppm type type shiny 0.1 box no 0.01 view 0 0 zoom 1.8 fsaa yes size 800 800
dump_modify viz adiam 1 1 adiam 2 3 acolor 1 turquoise acolor 2 royalblue backcolor white
The two variables n1_in, n2_in, along with the compute
sumcoor12, were added to the list of information printed during
the simulation. Additionally, images of the system will be created with
slightly less saturated colors than the default ones.
Finally, add the following lines to improved.md.lmp:
# 5) Run
velocity all create 1.0 49284 mom yes dist gaussian
fix mynve all nve
fix mylgv all langevin 1.0 1.0 0.1 10917 zero yes
timestep 0.005
run 300000
Here, there are a few more differences from the previous simulation.
First, the velocity create command assigns an initial velocity
to each atom. The initial velocity is chosen so that the average
initial temperature is equal to 1.0 temperature units. The additional
keywords ensure that no linear momentum (mom yes) is given to
the system and that the generated velocities are distributed according
to a Gaussian distribution. Another improvement is the zero
yes keyword in the Langevin thermostat, which ensures that the total
random force applied to the atoms is equal to zero. These steps are
important to prevent the system from starting to drift or move as a
whole.
Note
A bulk system with periodic boundary conditions is expected to remain in place. Accordingly, when computing the temperature from the kinetic energy, we use \(3N-3`\) degrees of freedom since there is no global translation. In a drifting system, some of the kinetic energy is due to the drift, which means the system itself cools down. In extreme cases, the system can freeze while its center of mass drifts very quickly. This phenomenon is sometimes referred to as the flying ice cube syndrome [20].
Run improved.md.lmp and observe the mixing of the two populations over time.

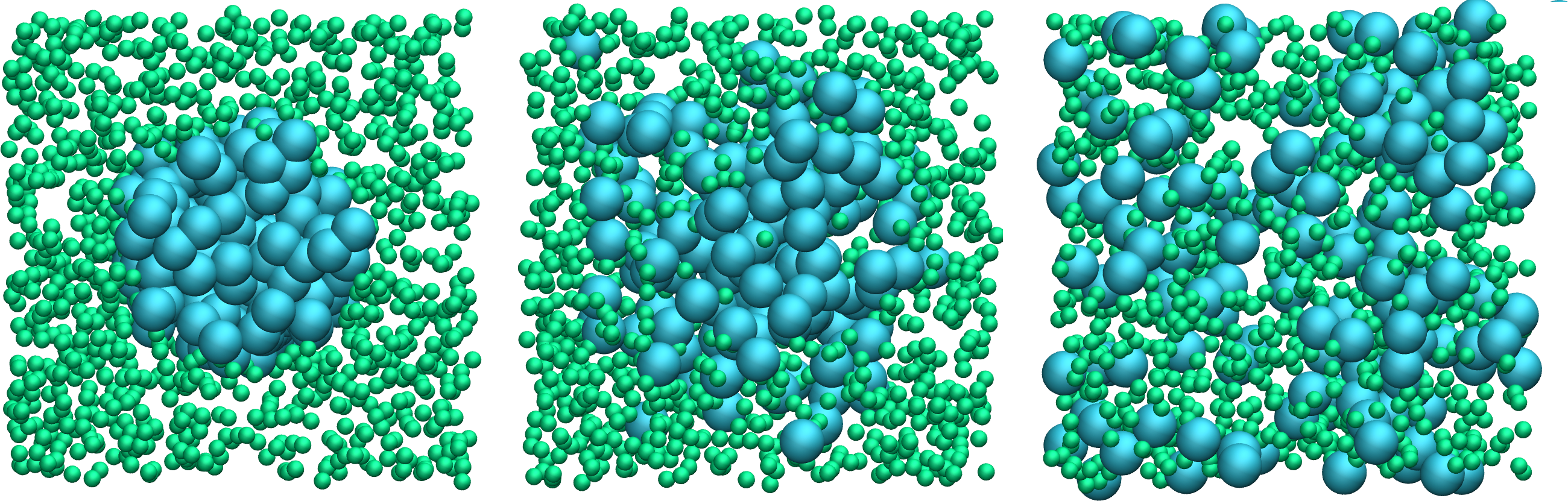
Figure: Evolution of the system during mixing. The three snapshots show respectively the system at \(t = 0\) (left panel), \(t = 75\) (middle panel), and \(t = 1500\) (right panel). The atoms of type 1 are represented as small green spheres and the atoms of type 2 as large cyan spheres.
From the variables n1_in and n2_in, you can track the number of atoms
in each region as a function of time (figure below, panel a). To view
their evolution, select the entries v_n1_in or v_n2_in in the Data
drop-down menu in the Charts window of LAMMPS–GUI.
In addition, as the mixing progresses, the average coordination number
between atoms of types 1 and 2 increases from about 0.01 to 0.04
(figure below, panel b). This indicates that, over time, more and
more particles of type 1 come into contact with particles of type 2, as
expected during mixing. This can be observed using the entry
c_sumcoor12 in the Charts drop-down menu.
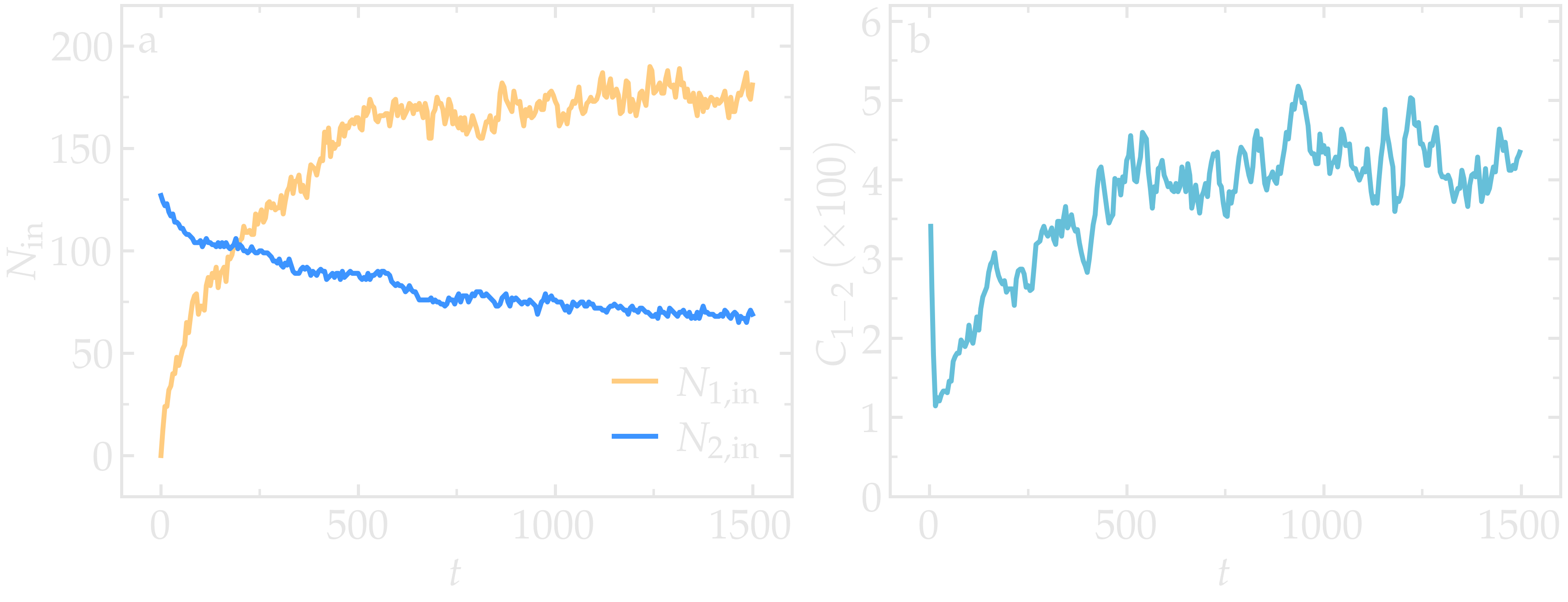
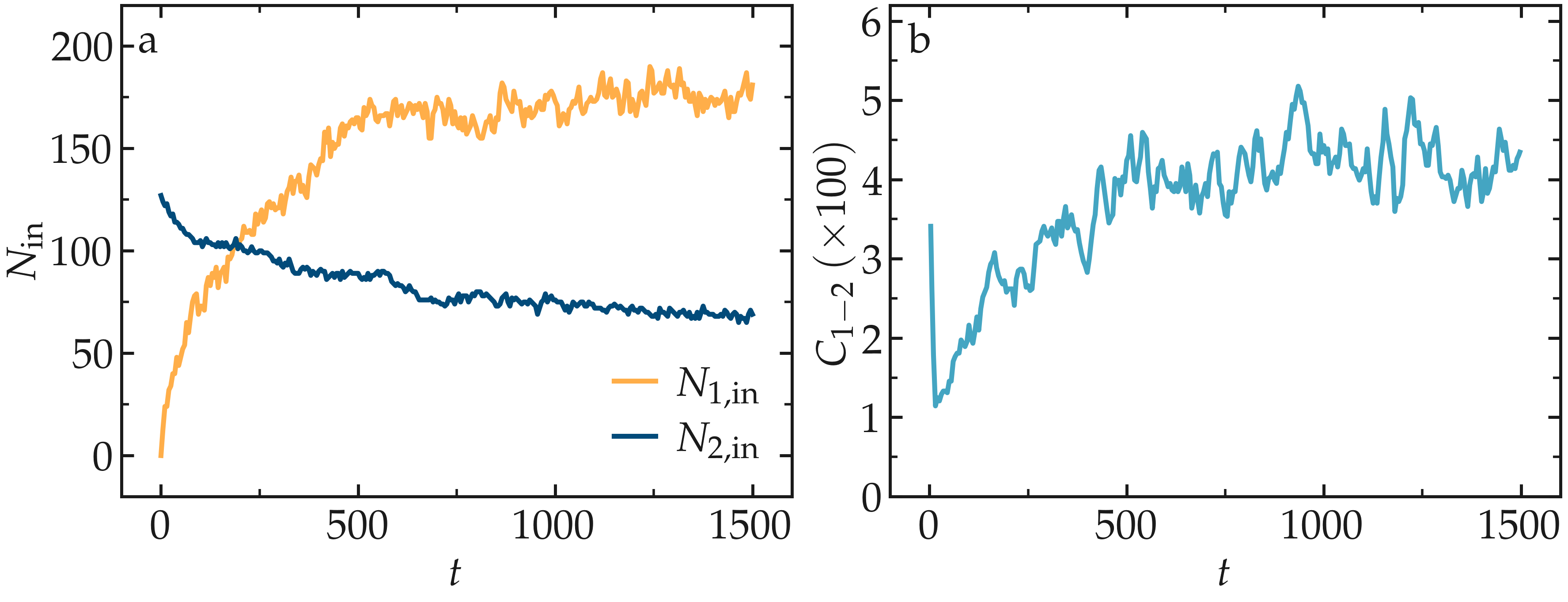
Figure: a) Evolution of the numbers \(N_\text{1, in}\) and \(N_\text{2, in}\) of atoms
of types 1 and 2, respectively, within the cyl_in region as functions
of time \(t\). b) Evolution of the coordination number \(C_{1-2}\)
(compute sumcoor12) between atoms of types 1 and 2.
Cite
You can access the input scripts and data files that are used in these tutorials from a dedicated GitHub repository. This repository also contains the full solutions to the exercises.
Going further with exercises¶
Experiments¶
Here are some suggestions for further experiments with this system that may lead to additional insights into how different systems are configured and how various features function:
Use a Nosé-Hoover thermostat (fix nvt) instead of a Langevin thermostat (fix nve + fix langevin).
Omit the energy minimization step before starting the MD simulation using either the Nosé-Hoover or the Langevin thermostat.
Apply a thermostat to only one type of atoms and observe the temperature for each type separately.
Append an NVE run (i.e., without any thermostat) and observe the energy levels.
If you are using LAMMPS-GUI
An useful experiment is coloring the atoms in the Slide Show according to an observable, such as their respective coordination numbers. To do this, replace the dump and dump_modify commands with the following lines:
variable coor12 atom (type==1)*(c_coor12)+(type==2)*-1
dump viz all image 1000 myimage-*.ppm v_coor12 type &
shiny 0.1 box no 0.01 view 0 0 zoom 1.8 fsaa yes size 800 800
dump_modify viz adiam 1 1 adiam 2 3 backcolor white &
amap -1 2 ca 0.0 4 min royalblue 0 turquoise 1 yellow max red
Run LAMMPS again. Atoms of type 1 are now colored based on the value of c_coor12, which is mapped continuously from turquoise to yellow and red for atoms with the highest coordination. In the definition of the variable v_coor12, atoms of type 2 are all assigned a value of -1, and will therefore always be colored their default blue.
Solve Lost atoms error¶
For this exercise, the following input script is provided:
units lj
dimension 3
atom_style atomic
pair_style lj/cut 2.5
boundary p p p
region simulation_box block -20 20 -20 20 -20 20
create_box 1 simulation_box
create_atoms 1 random 1000 341841 simulation_box
mass 1 1
pair_coeff 1 1 1.0 1.0
dump mydmp all atom 100 dump.lammpstrj
thermo 100
thermo_style custom step temp pe ke etotal press
fix mynve all nve
fix mylgv all langevin 1.0 1.0 0.1 1530917
timestep 0.005
run 10000
As it is, this input returns one of the most common error that you will encounter using LAMMPS:
ERROR: Lost atoms: original 1000 current 984
The goal of this exercise is to fix the Lost atoms error without using any other command than the ones already present. You can only play with the values of the parameters and/or replicate every command as many times as needed.
Note
This script is failing because particles are created randomly in space, some of them are likely overlapping, and no energy minimization is performed prior to start the molecular dynamics simulation.
Create a demixed dense phase¶
Starting from one of the input created in this tutorial, fine-tune the parameters such as particle numbers and interaction to create a simulation with the following properties:
the density in particles must be high,
both particles of type 1 and 2 must have the same size,
particles of type 1 and 2 must demix.


Figure: Snapshots taken at different times showing the particles of type 1 and type 2 progressively demixing and forming large demixed areas.
Hint
An easy way to create a dense phase is to allow the box dimensions to relax until the vacuum disappears. You can do that by replacing the fix nve with fix nph.
From atoms to molecules¶
Add a bond between particles of type 2 to create dumbbell molecules instead of single particles.


Figure: Dumbbell molecules made of 2 large spheres mixed with smaller particles (small spheres). See the corresponding video.
Similarly to the dumbbell molecules, create a small polymer, i.e. a long chain of particles linked by bonds and angles.

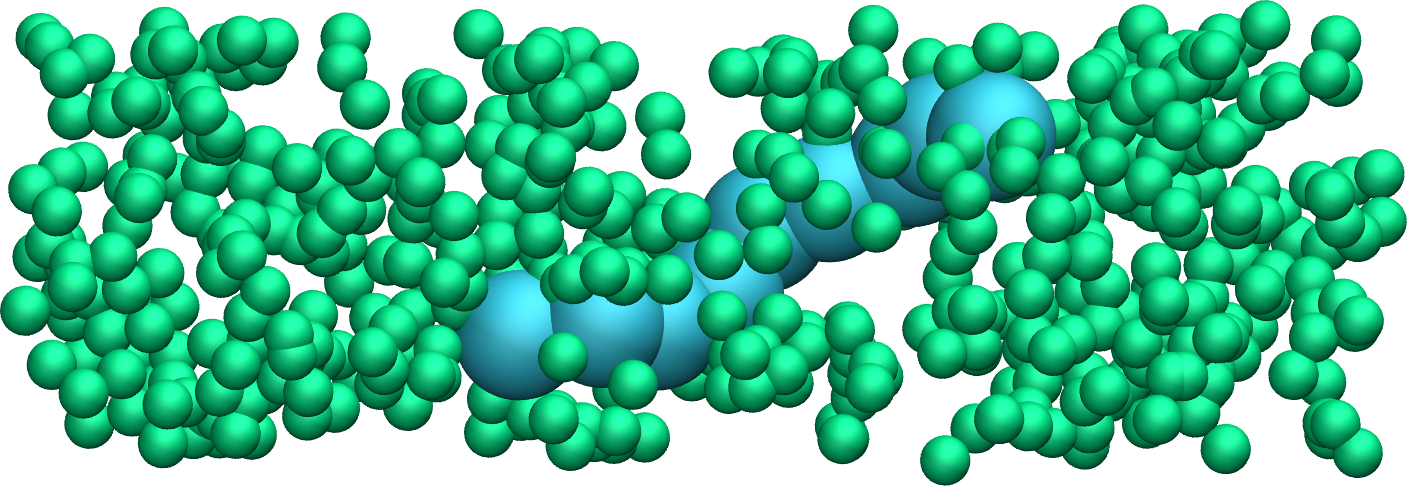
Figure: A single small polymer molecule made of 9 large spheres mixed with smaller particles. See the corresponding video.
Hints
Use a molecule template to easily insert as many atoms connected by bonds (i.e. molecules) as you want. A molecule template typically begins as follows:
2 atoms
1 bonds
Coords
1 0.5 0 0
2 -0.5 0 0
(...)
A bond section also needs to be added, see this page for details on the formatting of a molecule template.